What are Inhibitors?
Inhibitors play an important role in regulating the activities of enzymes.
Comparison of competitive and non-competitive inhibition of enzymes with reference to protein structure, the active site and allosteric site.
Inhibitors are compound that decrease the activity of an enzyme by binding to the enzyme and affecting the ability of the enzyme to bind to the substrate.
There are 2 types of inhibitors, competitive and non competitive inhibitors which act in different ways to reduce the activity of the enzyme.
Competitive Inhibitors
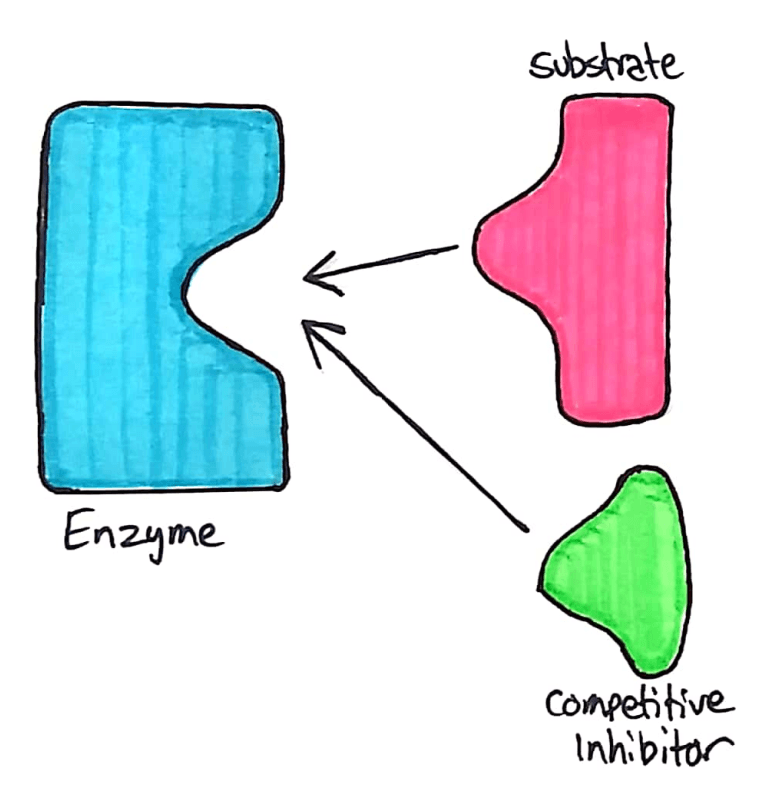
Competitive inhibitors compete with the substrate, and bind directly to the active site.
The competitive inhibitor acts similarly to the substrate. It binds to the enzyme’s active site through intermolecular forces, and temporarily stops the enzyme from reacting with any substrate. This reduces the speed at which an enzyme can break down substrate.
Understanding Competitive Inhibitors
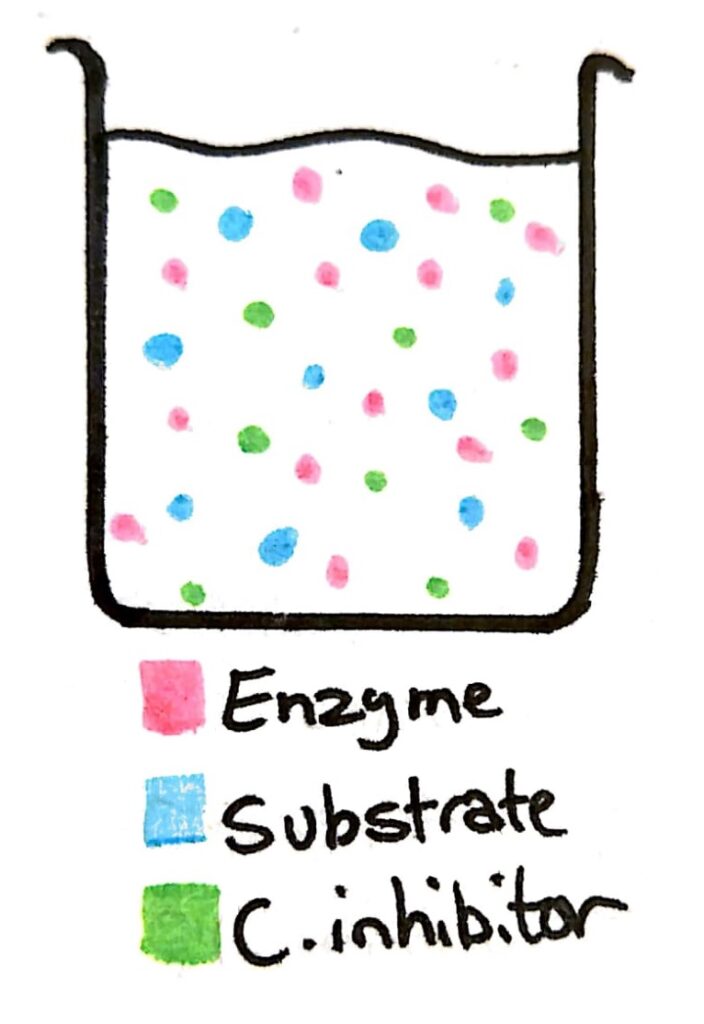
Let’s say we have a beaker with a concentration of enzyme, and concentrations of substrate and the competitive inhibitor that are similar.
The possibility of an enzyme colliding and binding with an enzyme or a competitive inhibitor is almost the same. Because 50% of the time the enzyme is binding with the inhibitor instead of the substrate, the rate of reaction is low
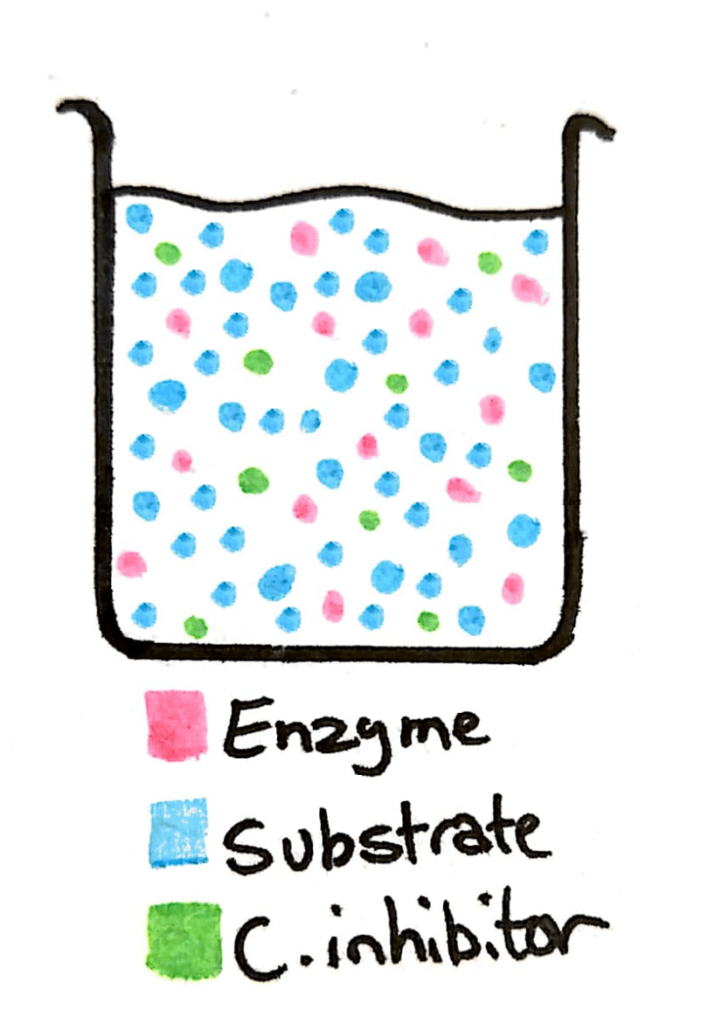
Now imagine in that same beaker you add a whole load more substrate. The concentration of the substrate just skyrocketed, and now there’s barely any competitive inhibitor in there compared to substrate.
This means that what was previously a 50/50 change between the inhibitor and the substrate is now a 90/10 or 99/1 ratio. The enzyme can easily reach it’s maximum rate of reaction.
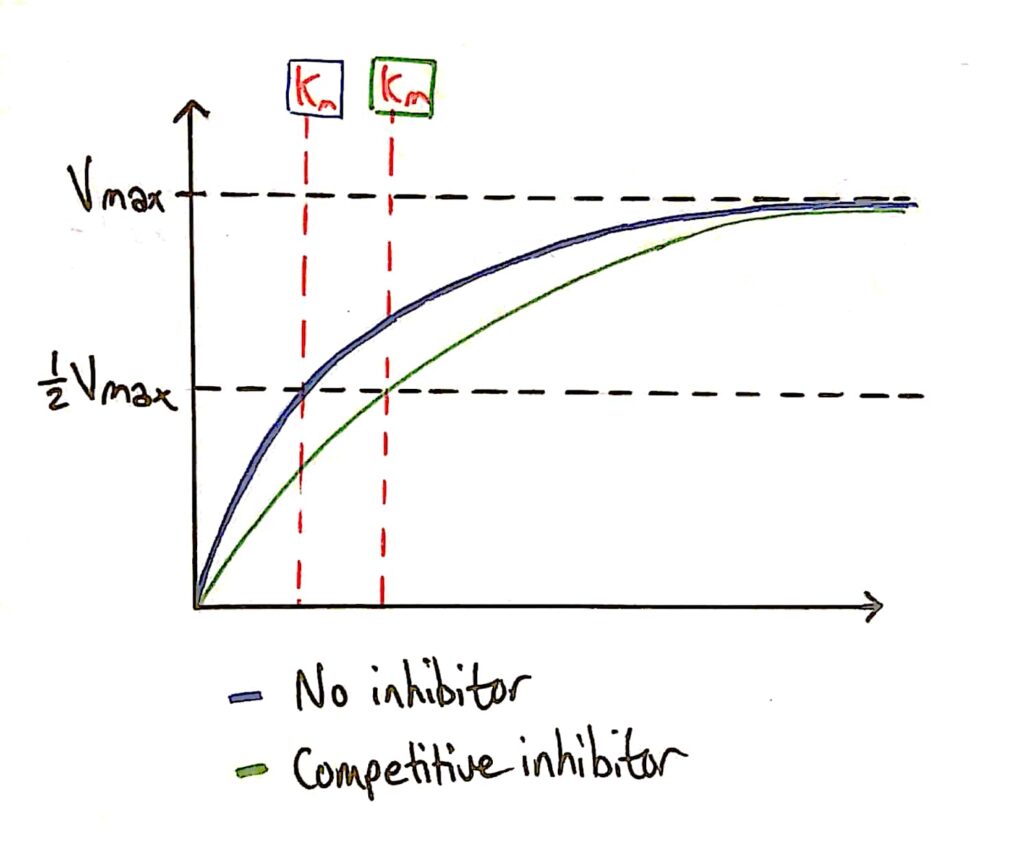
Enzyme Activity - Competitive Inhibitors
As I said earlier, the addition of a competitive inhibitor does not effect the maximum rate of reaction of the enzyme because you can just add more substrate.
The Km (Michaelis constant) decreases however because the competitive inhibitor affects how well the enzyme can bind to the substrate (C. inhibitor gets in the way).
Non-Competitive Inhibitors
Non competitive inhibitors, as the name suggests, does not compete with the substrate to bind to the active site. Instead, competitive inhibitors bind to an allosteric site.
The allosteric site on an enzyme is a specific part of the enzyme where a non competitive inhibitor binds. When the inhibitor binds to the allosteric site, the active site is deformed and the enzyme no longer works.
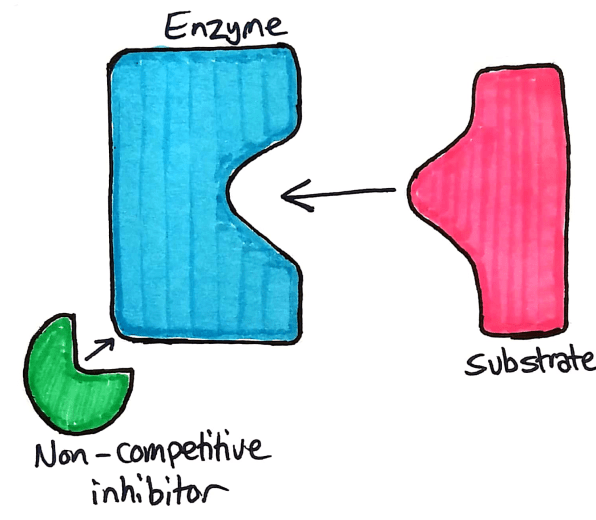
Understanding Non-competitive Inhibitors
If you add non-competitive inhibitors to a beaker of enzymes, the inhibitors bind permanently to the enzymes. The enzymes become completely useless because their active site has been deformed.
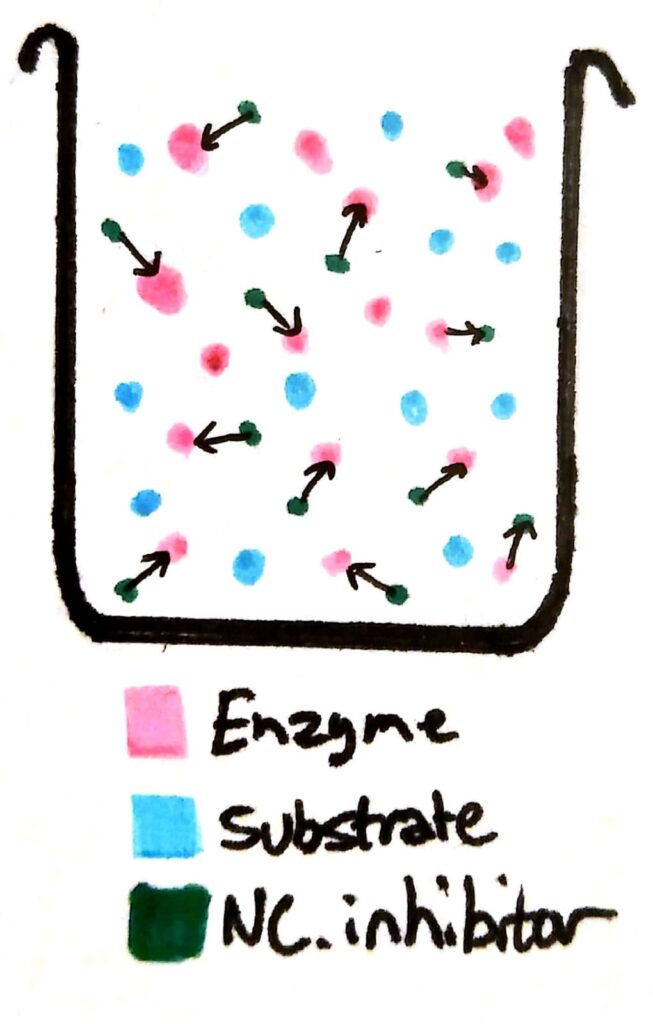

Now, a whole load of the enzymes have been essentially ‘deactivated’. Compared to a beaker with no non-competitive inhibitors, the maximum rate of reaction will be less no matter how much you increase the substrate concentration, because there’s barely any working enzymes left in solution.
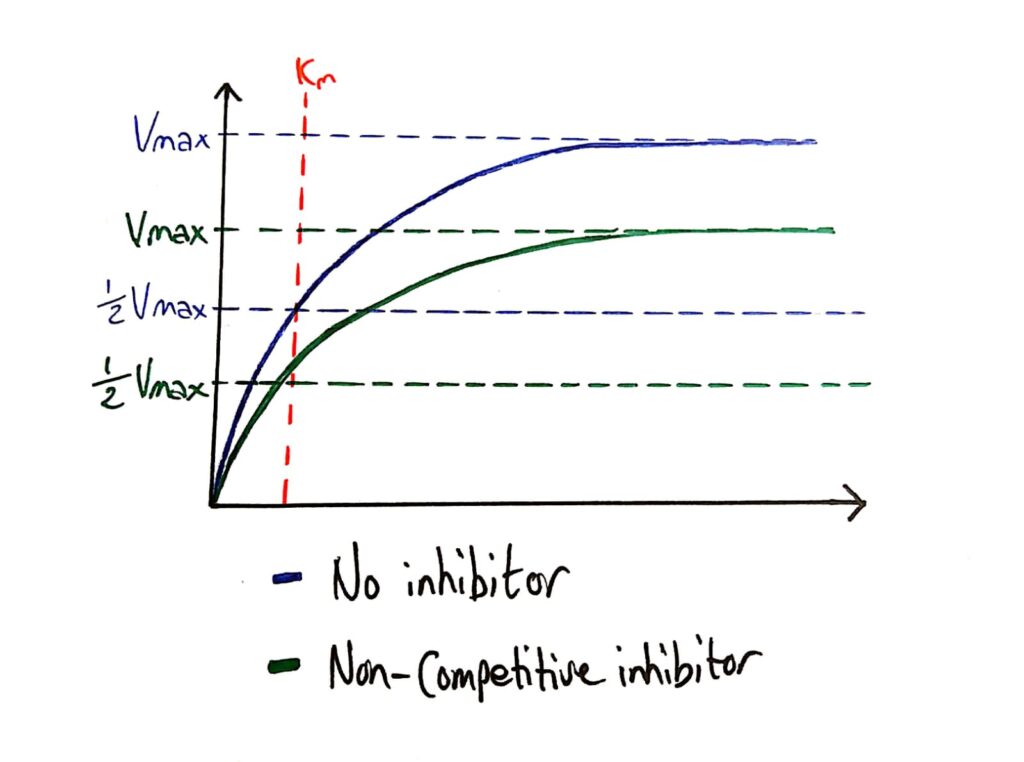
Enzyme Activity - Non-competitive Inhibitors
Because non-competitive inhibitors essentially deactivate enzymes, they lower the maximum rate of reaction of the enzyme.
What’s cool is that the Km (Michaelis constant) is not affected by non-competitive inhibitors because non-competitive inhibitors do not affect how well the enzyme bonds to the substrate, it just ‘deactivates’ them.
Examples of Non Competitive Inhibitors
- Heavy metal ions
- Mercury (Hg2+)
- Lead (Pb2+)
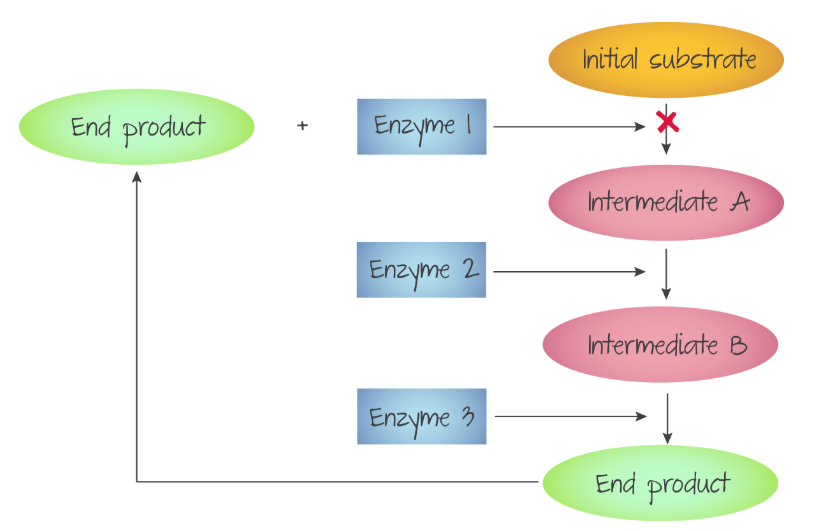
Product Inhibition
In some reactions, the product of the reaction between the enzyme and substrate can be an inhibitor. Since metabolic processes often have multiple enzyme catalysed steps, the end product of the whole process sometimes produces compounds that inhibit one of the enzyme catalysed steps.
Summary
Competitive Inhibitor
Competitive inhibitors compete with the substrate
- Bind to the active site
- Binds non-permanently through weak IMFs
- Reduces the Km of the enzyme
- Does not affect the maximum rate
Non-competitive Inhibitor
Non Competitive inhibitors bind to an enzyme and essentially ‘deactivate‘ it
- Bind to the allosteric site
- Binds permanently through covalent bonds
- Does not affect the Km of the enzyme
- Reduces the maximum rate
PROTEIN ASSAYS
TESTING FOR PROTEIN
A qualitative test for proteins that you’ve probably done before is the Biuret’s test. A volume of Biuret’s reagent is added to the sample to be analysed.

The Biuret’s test can be used to measure the concentration of a solution of proteins using colourimetry because of its purple colour.
COLOURIMETRY
Colourimetry is the determination of the concentration of a solution based on how much light it absorbs. The amount of light that is absorbed can be measured using a colourimeter, and an absorbance value can be calculated. This technique works for any solution as long as it’s not colourless.
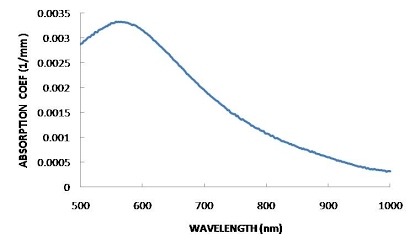
FINDING THE OPTIMUM WAVELENGTH
In order to find the wavelength that is absorbed most by the solution, an absorption spectrum should be plotted. This is basically an absorbance against frequency/wavelength graph that will show how well each wavelength is absorbed. The wavelength with the highest absorption should be chosen
HOW IT WORKS
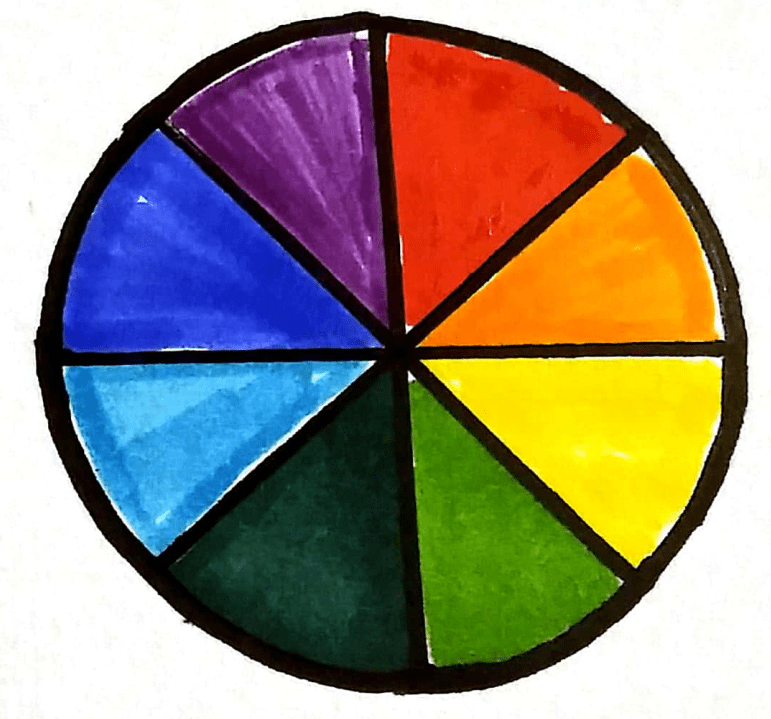
The complementary colour wheel
Coloured solutions absorb certain wavelengths of light depending on their colour. If we know how much light is absorbed by a known concentration value, we can estimate the concentration of unknown samples by comparing the amount of light it absorbs compared to the known sample
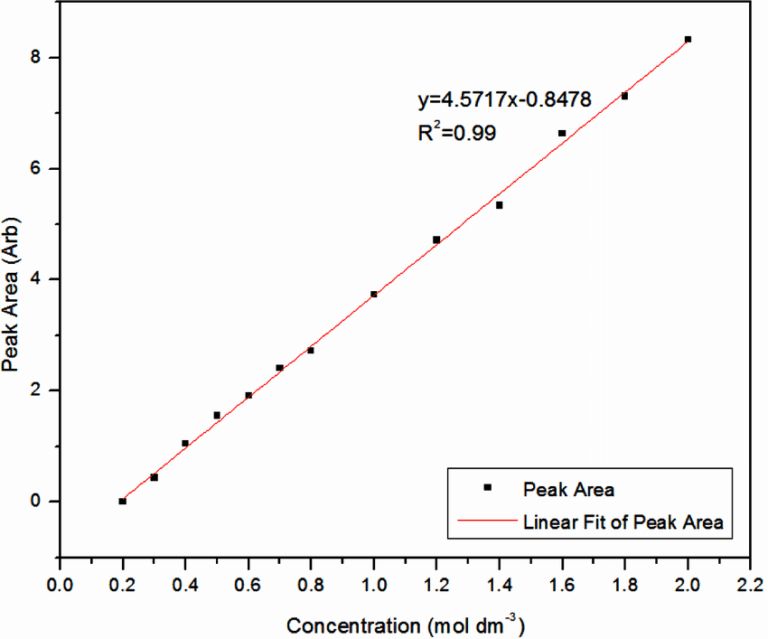
CALIBRATION CURVES
Calibration curves are absorbance-concentration graphs that are used to compare unknown samples to. Absorbance values for samples of known concentration are taken and plotted on a graph, and a line of best fit is drawn. From this graph, any unknown concentrations can be determined by finding their absorbance values on the line of best fit and tracing back to the concentration.
METHODOLOGY (for finding protein concentration)
- Use the absorbance values of known concentrations of the protein solution to plot a calibration curve
- Add Biuret solution to the unknown protein sample and test absorbance in a colourimeter.
- Use absorbance value and line of best fit to find concentration value.
THE BEER-LAMBERT LAW
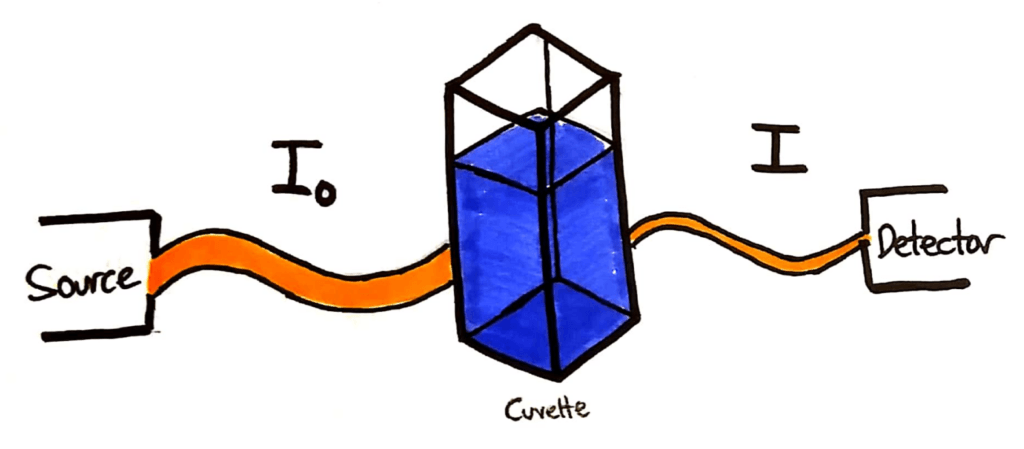
A typical colourimetry setup. An incident beam, a cuvette of solution and a detector.
The light source will shine a beam of light through the cuvette of sample. The intensity of that light beam is measured, and given the symbol I0. The beam then passes through the sample in the cuvette, and a portion of the light is absorbed. The remaining light beam is detected at the detector, and it’s intensity is given the symbol I.
THE MATHS
In the data book, you’re given the following formula:

What they don’t tell you in this formula, is that both sides equal A, the Absorbance of the solution.
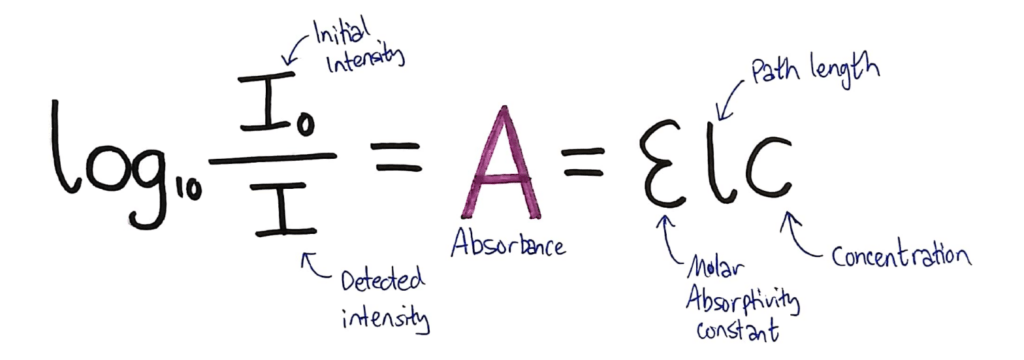
Above, I’ve added the absorbance to the formula and annotated the formula to show what each variable is.
THE VARIABLES
- The intensity of the initial beam
- The intensity detected after the beam has passed through the solution
- The absorbance value for the solution
- The molar absorptivity constant which is different for every substance
- The path length. This is how much solution the light has to travel through. In a standard colourimetry experiment standard cuvettes are used which are 1cm, so it usually cancels.
- The concentration of the solution, which you’re usually trying to find.